HMMER is a bioinformatics tool for analyzing gene and protein sequences, especially when you need to measure similarity between sequence sets. The latest release in the main line is 3.1, but the official site only provides Linux builds now. The Windows version has effectively stopped there. That may sound like one more example of sequence-analysis software being happiest on Linux, although the official documentation also notes that HMMER4 has been under development since 2011 and progress has been slow, which likely explains why the Windows side was never kept current.
For anyone who only needs a workable Windows setup, the older Windows build can still be used.
Installing HMMER on Windows
Downloading the last Windows release
If setting up a Linux virtual machine just for HMMER feels like overkill, an easier option is to look through the archived releases. Following the download trail for the Linux packages leads to the full software archive, where the final Windows version can still be found: HMMER 3.0.

After downloading and extracting it, you will not see a clickable .exe application. That is normal. HMMER is meant to be run from the command line.
Adding HMMER to the system path
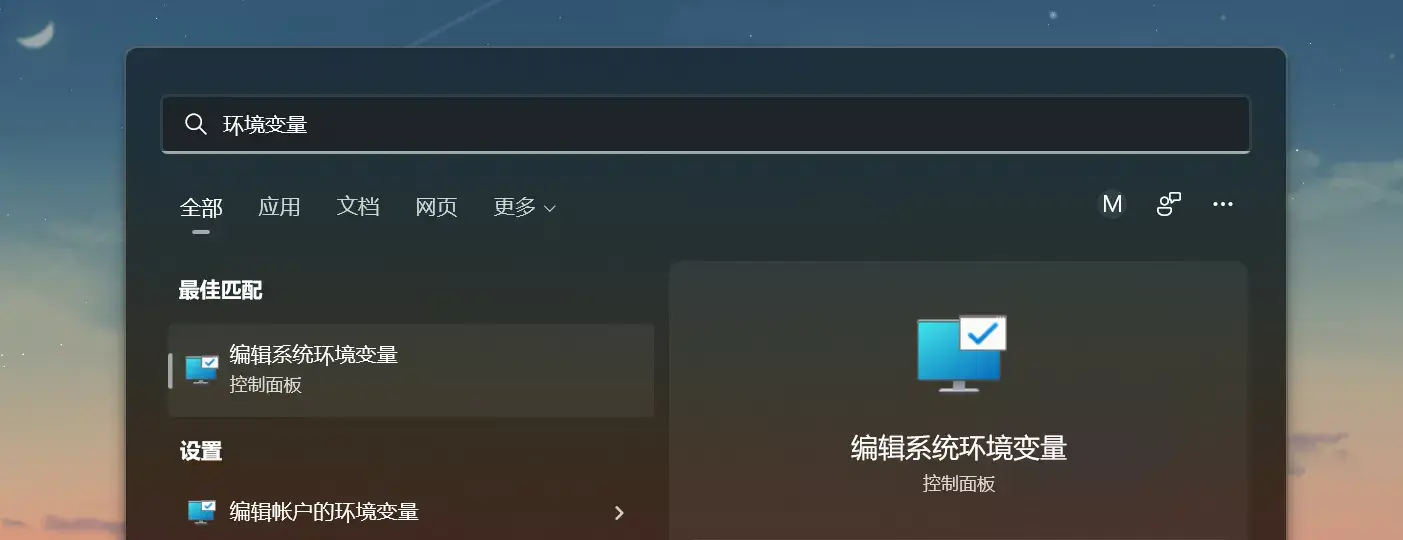
On Windows 10, open Control Panel, search for environment variables, click Edit the system environment variables, and then choose Environment Variables.

On Windows 11, open the Start menu and type environment variables into the search bar.
Open the first matching result.
Under System variables, find Path and click Edit.

Click New and enter the folder path where HMMER is stored.

If you are not sure what path to use, open the folder containing HMMER, copy the text from the File Explorer address bar, and paste it into the variable editor.

After that, confirm the changes and close the dialogs.
Testing whether the installation works
To check that HMMER is available from the command line, use either CMD or Windows PowerShell.
To open CMD, press Windows + R, type cmd, and confirm.

To open Windows PowerShell, right-click the Start menu and choose Windows PowerShell.

Then enter:
hmmscan -h
Copy and paste is safer than typing by hand. If a help screen appears, HMMER has been installed correctly.

A practical HMMER workflow
The two commands used here are hmmbuild and hmmsearch.
hmmbuild: creates an HMM profile from an alignmenthmmsearch: searches a sequence set against that HMM profile
The example below uses a MADS-box gene from Jatropha curcas.
1. Find the Pfam ID for the conserved domain
To compare sequences with HMMER, you first need the hidden Markov model corresponding to the gene family or conserved domain of interest. In practice, that means identifying the relevant Pfam entry.
One way is to look it up in published work. Another is to identify it directly through NCBI.
Go to the NCBI Protein database and search using a gene keyword plus the species name in Latin or formal English. For example:
MADS-box Jatropha curcas

Open any suitable protein record from the results, then click Identify Conserved Domains on the right side to analyze its conserved domains.

In the domain results, the target protein is shown hitting the K-box family, and the Pfam ID is listed there directly.

2. Download the conserved-domain alignment
Click the Pfam ID from the result above to open the conserved domain details, then follow the source link to Pfam.

On the Pfam page, open Alignments, choose Stockholm format, and click generate to download the multiple sequence alignment. The downloaded file will be a .txt file.

3. Download the species protein sequences
Next, download the protein data for the species you want to search against. In this example, that is the Jatropha curcas genome protein set.
Go to the NCBI FTP site, enter genomes, and use Ctrl+F in the browser to search for the species name. Since the Latin name is Jatropha curcas, searching for Jatropha is enough to locate it.

In the next directory, choose protein and download protein.fa.gz. After extracting it, you will get protein.fa.
4. Build the model and run the search
Copy both downloaded files into the HMMER folder. They can be placed elsewhere too, but keeping them in the same working directory makes the commands simpler.
Then open the command prompt.
By default, Command Prompt starts in a directory like C:\Users\username>. You need to switch to the directory where HMMER is located before continuing.
If HMMER is not on drive C, switch drives first. For example, if it is on drive D:
D:
Then change into the HMMER folder:
cd HMMER安装位置
For example:
cd D:\hmmer
If that fails, the path may contain Chinese or other non-English characters. In that case, wrap the path in single quotes:
cd 'D:\假装有中文\hmmer'
Now use hmmbuild to convert the conserved-domain alignment into an HMM profile. If the downloaded alignment file is named PF01486_seed.txt, enter:
hmmbuild hmm文件 需要转换的文件
For example:
hmmbuild PF01486.hmm PF01486_seed.txt
Once the .hmm file has been created, compare it against the Jatropha curcas protein sequences with hmmsearch:
hmmsearch PF01486.hmm protein.fa > PF01486.out

When the command finishes, it will generate PF01486.out. Right-click that file and open it in Notepad.

The alignment results can then be inspected directly in the output file.